Overview of the models¶
As the following figure shows, the extrinsic apoptosis pathway can be considered to have roughly three major regulatory focal points, or “modules”: [Albeck2008]
- Activation of initiator caspases of Bid by receptor ligation (“Receptor to Bid” in the diagram)
- Mitochondrial outer membrane permeabilization (MOMP)
- Effector caspase activation and substrate cleavage (“Pore to PARP cleavage” in the diagram)
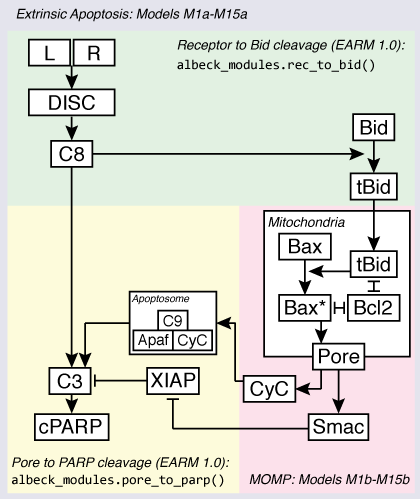
The models in EARM are focused on exploring alternate hypotheses for the regulation of MOMP by Bcl-2 family proteins, both as an isolated module and in the overall context of the extrinsic apoptosis pathway.
Each hypothesis for MOMP regulation by Bcl-2 proteins in EARM thus has two models associated with it: a “MOMP-only” model that can be used to study the properties of a Bcl-2 reaction topology as an isolated module, and a “full apoptosis” form, in which the different MOMP models are embedded in the full extrinsic apoptosis pathway which begins with TRAIL or FasL stimulation. EARM contains 15 alternative Bcl-2 topologies for MOMP; thus there are 15 Bcl-2 topologies x two versions = 30 models, enumerated M1a, M1b, ..., M15a, M15b. The “a” suffix denotes the full apoptosis model for a given Bcl-2 topology, while the “b” suffix denotes the corresponding MOMP-only model.
For the full apoptosis models, the upstream and downstream pathway components and reaction topologies are re-used from the previously published EARM 1.0 [Albeck2008].
The models in EARM¶
Below is a list of the 15 alternative Bcl-2 reaction topologies incorporated into EARM. More detailed descriptions of each model, along with the source code, are found in Implementation details (code and documentation).
- M1a/b: EARM 2.0, Embedded [Lopez2013]
- M2a/b: EARM 2.0, Direct [Lopez2013]
- M3a/b: EARM 2.0, Indirect [Lopez2013]
- M4a/b: “Minimal Model” (Figure 11b) from Albeck et al. (2008) [Albeck2008]
- M5a/b: “Model B + Bax multimerization” (Figure 11c) from Albeck et al. (2008) [Albeck2008]
- M6a/b: “Model C + mitochondrial transport” (Figure 11d) from Albeck et al. (2008) [Albeck2008]
- M7a/b: “Current model” (Figure 11e) from Albeck et al. (2008) [Albeck2008]
- M8a/b: “Current model + cooperativity” (Figure 11f) from Albeck et al. (2008) [Albeck2008]
- M9a/b: Deterministic model from Chen et al. (2007), Biophysical Journal [Chen2007biophysj]
- M10a/b: Indirect model from Chen et al. (2007), FEBS Letters [Chen2007febs]
- M11a/b: Direct model from Chen et al. (2007), FEBS Letters [Chen2007febs]
- M12a/b: Direct model from Cui et al. (2008) [Cui2008]
- M13a/b: Direct model 1 from Cui et al. (2008) [Cui2008]
- M14a/b: Direct model 2 from Cui et al. (2008) [Cui2008]
- M15a/b: Model incorporating Bad phosphorylation from Howells et al. (2011) [Howells2011]
How the model code is organized¶
For each of the 30 models, there is a corresponding .py file containing
the model definition. This way any model can be imported using the
straightforward syntax (for example, for model M1a):
from earm.lopez_embedded import model
However, the Python files for each individual model in general do not contain
much code–they mainly call functions from other modules. For example,
here is the source code for the file earm/lopez_embedded.py, which
implements model M1a:
"""
Model M1a: Extrinsic apoptosis model with expanded, "embedded together"
model of MOMP.
"""
from pysb import *
from earm import shared
from earm import lopez_modules
from earm import albeck_modules
Model()
# Declare monomers
albeck_modules.ligand_to_c8_monomers()
lopez_modules.momp_monomers()
albeck_modules.apaf1_to_parp_monomers()
# Generate the upstream and downstream sections
albeck_modules.rec_to_bid()
albeck_modules.pore_to_parp()
# The specific MOMP model to use
lopez_modules.embedded()
# Declare observables
shared.observables()
As this example shows, the model file calls a series of macros that declare the
monomers (earm.albeck_modules.ligand_to_c8_monomers(),
earm.lopez_modules.momp_monomers(), and
earm.albeck_modules.apaf1_to_parp_monomers()), then calls the macros
implementing the upstream and downstream pathway elements
(earm.albeck_modules.rec_to_bid() and
earm.albeck_modules.pore_to_parp()), and finally calls the macro for
the specific Bcl-2 topology involved: earm.lopez_modules.embedded().
Since the observables for all of the full apoptosis model variants is the same,
these are declared in the final macro that is called,
earm.shared.observables().
All of the model .py files follow this pattern, calling a handful of macros
to declare monomers and observables and select implementations of different
pathway modules.
Note that the .py model files for the full-apoptosis models (M1a - M15a)
are found in the top-level earm module, but the files for the MOMP-only
models (M1b - M15b) are found in the submodule earm.mito.
The documentation for all model files (with links to source code) can be found at the following links:
The details of the various macro implementations are found in the following four files:
MOMP module “boundaries”¶
In the interest of consistency, all of the models have been defined with the same boundaries in terms of their position in the overall extrinsic apoptosis pathway: they are all triggered by the addition of an active BH3-only species (e.g., tBid) as their most “upstream” event, and they all result in the release in one or more mitochondrial substances (e.g. Cytochrome C and/or Smac) as their most downstream event. This represents a compromise between the approach of the MOMP models described in Albeck et al (in which caspase-8, rather than tBid, served as the input) and the models of the Shen group, in which active Bax or Bax pores, rather than Cytochrome C or Smac, served as the output.
While these interface boundaries represent the default condition, they can be modified by passing parameters in to the module macro. For example, by setting do_pore_transport=False in the call to one of the Shen models, the Cytochrome C and Smac release reactions are not added, and the models can be directly compared to their originally published versions. Similarly, the upstream caspase-8/Bid reactions can be added to the Albeck MOMP models to make them consistent with their published versions.
Since our purpose in using these models is primarily to embed them in a common pathway context, rather than to reproduce previous results for posterity, our conclusion in working with them was that it is better to have a consistent interface by default and reproduce published results by modifying the model rather than implement the model as published by default and then have to specifically modify each one separately to fit the pathway context appropriately.
How to use the models¶
To import a model use the syntax:
from earm.lopez_embedded import model
That’s it. You now have a model object that you can query, simulate, perform
parameter estimation on, etc. If you wanted the MOMP-only version, which is
in the sub-module mito, simply run:
from earm.mito.lopez_embedded import model
If you want to work with multiple models at the same time (e.g., to compare them), you can write:
from earm.chen2007_indirect import model as indirect
from earm.chen2007_direct import model as direct
For more information on the kinds of analysis you can do using PySB models, see the PySB documentation.
Parameter values¶
Parameter values (both rate constants and initial protein concentrations) are embedded directly in the model code rather than in a separate table or file. The values in the model definition represent estimates or nominal values and can be easily overridden using values obtained (for example) by measurement or parameter estimation algorithms. We do not maintain a separate list or table of parameter values, as we have found that the clearest description of the meaning of a rate parameter is the macro or rule statement in which it is embedded.
If desired, lists of all model parameters can be obtained via the parameters instance variable of the model object, i.e.:
model.parameters
A list of all parameter names can be obtained using the list comprehension:
[p.name for p in model.parameters]
The code is meant to be read!¶
As much as possible, we have attempted to make the code for models themselves transparent and well-documented. The documentation for each model topology has been embedded inline in the model source code, and the documentation provided in the Implementation details (code and documentation) section of the documentation is drawn directly from this source.
Moreover, the models have been written using a high-level vocabulary of frequently re-used macros and motifs, with the aim of revealing broad similarities and differences between models. The models thus consist of statements such as:
translocate_tBid_Bax_BclXL()
catalyze(Bid(state='T'), Bax(state='M'), Bax(state='A'), klist)
which can be read as saying that “tBid, Bax and BclXL translocate [to the mitochondrial membrane], and tBid catalyzes Bax from a Mitochondrial (but inactive) state to an Active state.” Understanding the precise mechanisms of these macros (as expressed in terms of rules and reactions) takes some familiarity with their implementation, but as there is a fairly limited set of macros, this should hopefully not present a significant barrier.